# Data Upload
With the SEQ Platform, you can directly upload your files or use the cloud browser to browse and select the files pre-uploaded to your account.
# Direct Data Upload
Click on the "Upload" button at your homepage and select "Somatic" option. Here, you can select FASTQ or VCF options.
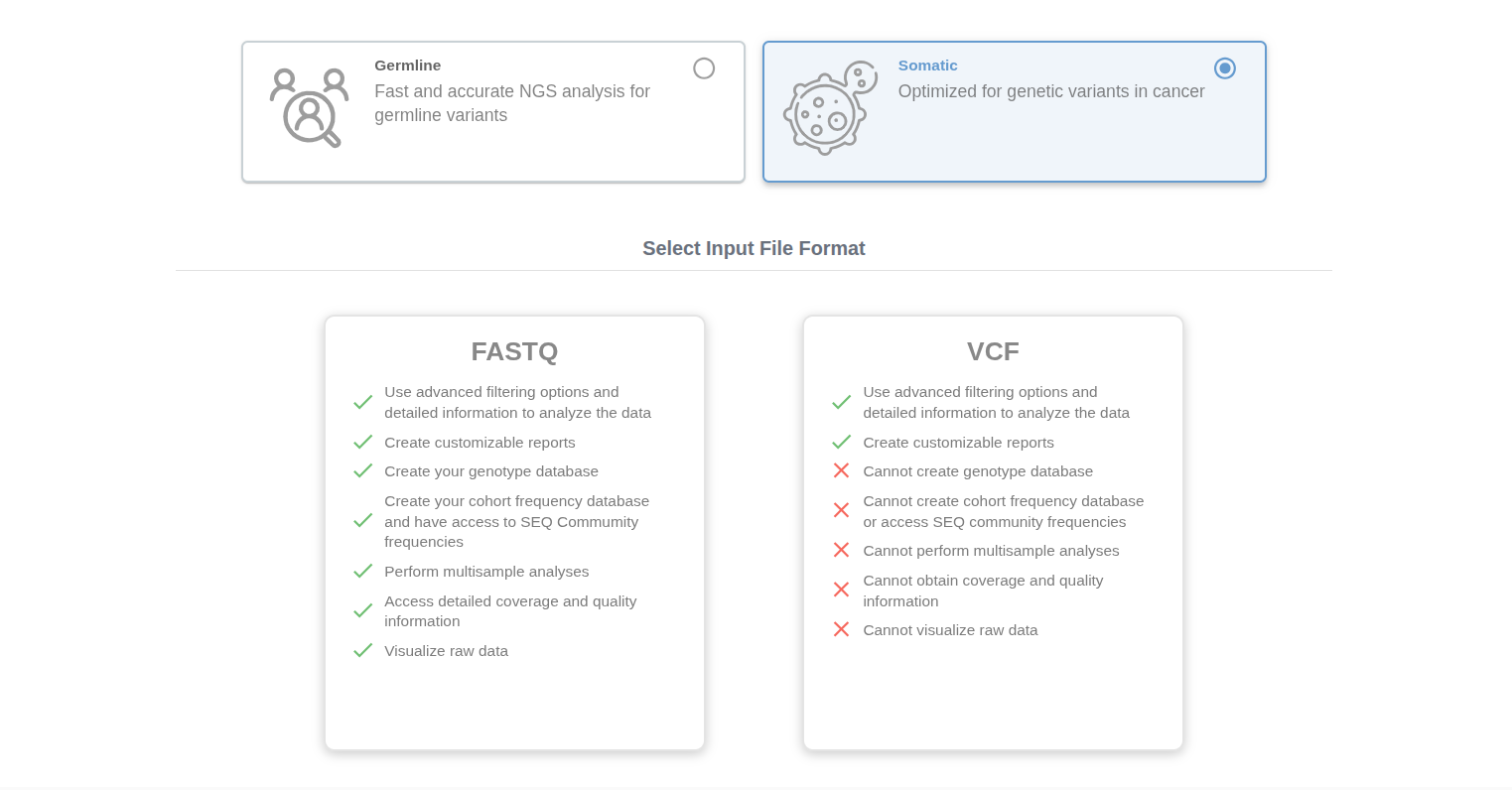
# FASTQ Upload
# Run Selection
You can upload your samples to a new run by selecting “Create New Run” under “Run Name” and giving it a name on the “Name For New Run” field. You can also upload your samples to an existing run by selecting the previous run from the dropdown menu.
# Diagnostics
Next, you can add the diagnostic information including cancer type and the biomarker status.

# Cancer Type
- (Mandatory) Cancer Type (e.g. Breast Cancer)
- (Optional) Advanced Solid Tumor: Select this option if the sample or case involves a late-stage or metastatic solid tumor, regardless of the specific cancer type.
# Biomarkers
- (Optional) TMB status: tumor mutation burden status (high | intermediate | low).The specified TMB status will be used in associating relevant clinical trials and drugs.
- (Optional) MSI status: Microsatellite instability status ( high | low | stable ). The specified MSI status is displayed in the report and used in associating relevant clinical trials and drugs.
- (Optional) HRD status: Homologous Recombination Deficiency status (high | low). The HRD status will be used in associating relevant clinical trials and drugs.
- (Optional) TILs status: Tumor Infiltrating Lymphocytes status (high | low). The TILs status will be used in associating relevant clinical trials and drugs.
- (Optional) PD-L1 status: PD-L1 (CD274) expression status (positive | negative). The PD-L1 (CD274) status will be used in associating relevant clinical trials and drugs.
- (Optional) HER2 status: HER2 (ERBB2) expression status (positive | negative). HER2 (ERBB2) status will be used in associating relevant clinical trials and drugs.
# Choose the Technology Type
Choose the next-generation sequencing machine associated with the samples. If you do not know which sequencing platform is used, you can select the "Unknown" option. Mixing different technologies in one run is not permitted.
# Choose the kit type
The SEQ platform has hundreds of different kits predefined in the system. A new kit can be defined with a set of target coordinates and a list of targeted genes. The Addition of a new kit typically takes one business day. For kit requests, please contact us through support@genomize.com.
Every kit is associated with a standardized analysis version in SEQ. Probe-based kits, primer-based kits, Illumina & MGI technology, ION torrent technology, germline analysis, or somatic analyses all have a preset analysis version.
# Select the files to upload
Click the "Browse" button under the “File” to upload all the files you want to analyze. Make sure that you upload both read files for paired-end reads. See the table below for the supported input file types:
| File Types | Batch Sample Upload | RNA-Seq Upload | |
|---|---|---|---|
| Illumina | .fastq.gz, .fq.gz | Supported | Supported |
| MGI | .fastq.gz, .fq.gz | Supported | Supported |
| Element Biosciences | .fastq.gz, .fq.gz | Supported | Supported |
| GeneMind | .fastq.gz, .fq.gz | Supported | Supported |
| Onso (PacBio) | .fastq.gz, .fq.gz | Supported | Supported |
| Salus | .fastq.gz, .fq.gz | Supported | Supported |
If you chose Tumor/Normalanalysis pipeline, Normal Files selection will be active. Using these fields, you can select the matched normal file for your tumor samples.
Filename formatting is explained below. Formatting rules apply to normal and tumor samples separately. Matching between the tumor and normal samples is not checked. System will assume correct matching tumor and normal files are selected by the user.
# Filename formatting for batch upload
# Illumina
You can upload multiple samples with two or more files. For batch uploads, we only support filenames following the “Illumina naming convention” e.g.
NA10831_ATCACG_L002_R1_001.fastq.gz
NA10831_ATCACG_L002_R2_001.fastq.gz
The filenames from Illumina platform are handled as below:
<name_field1>_<name_field2>_<lane_#>_<read_#>_<always_001>.fastq.gz
Name field 1, name field 2, lane #, and read # are used to match the corresponding files correctly.
You can alter the name fields 1 and 2 without using space, punctuation, or underscore (_) characters.
Following this naming convention, you can upload multiple samples (each with multiple fastq.gz files).
The --no-lane-splitting parameter must not be used during file conversion with bcl2fastq, DRAGEN BCL Convert, or similar tools.
Lane identifiers are required to remain present in FASTQ file names for correct file matching. Please refer to the tools’ documentations for details.
Please check the number of samples and matched files on the confirmation screen.
# MGI
You can upload multiple samples with two or more files. For batch uploads, we only support filenames following the “MGI naming convention” e.g.
V12345678_L01_16_1.fastq.gz
V12345678_L01_16_2.fastq.gz
The filenames from MGI platform are handled as below:
<flowcell_id>_<lane_#>_<barcode>_<read_#>.fq.gz
Flowcell ID, lane #, barcode, and read # are used to match the corresponding files correctly.
You can alter the flowcell ID field without using space, punctuation or underscore (_) characters.
If you have used more than one barcode for the same sample, you need to rename the file as follows:
Original file names:
sample1234_L01_16_1.fq.gz
sample1234_L01_16_2.fq.gz
sample1234_L01_17_1.fq.gz
sample1234_L01_17_2.fq.gz
Altered file names:
sample1234_L01_16_1.fq.gz
sample1234_L01_16_2.fq.gz
sample1234_L02_16_1.fq.gz
sample1234_L02_16_2.fq.gz
In this example, barcode numbers of the last two files are changed to 16, and their lane numbers are increased by 1.
Please check the number of samples and matched files on the confirmation screen.
# Element Biosciences
You can upload multiple samples with two or more files. For batch uploads, we only support filenames following the “Element Biosciences naming convention” e.g.
SampleName_S1_L001_R1_001.fastq.gz
SampleName_S1_L001_R1_001.fastq.gz
The filenames from Element platforms are handled as below:
<name_field1>_<name_field2>_<lane_#>_<read_#>_<always_001>.fastq.gz
Name field 1, name field 2, lane #, and read # are used to match the corresponding files correctly.
You can alter the name fields 1 and 2 without using space, punctuation, or underscore (_) characters.
Following this naming convention, you can upload multiple samples (each with multiple fastq.gz files).
Please make sure the Lane identifiers are present in the file names as they are required for correct file matching.
You can use --legacy-fastq parameter in Bases2Fastq to produce files with names adhering to the above format.
Please refer to the Bases2Fastq documentation for details.
Please check the number of samples and matched files on the confirmation screen.
# GeneMind
You can upload multiple samples with two or more files. For batch uploads, we only support filenames following the “GeneMind naming convention” e.g.
SampleName_R1.fq.gz
SampleName_R2.fq.gz
The filenames from GeneMind platform are handled as below:
<name_field>_<read_#>.fq.gz
Name field and read # are used to match the files correctly.
You can alter the name field without using space, punctuation and underscore (_) characters.
Following this naming convention, you can upload multiple samples (each with multiple fq.gz files).
Please check the number of samples and matched files on the confirmation screen.
# Salus
You can upload multiple samples with two or more files. For batch uploads, we only support filenames following the “Salus naming convention” e.g.
ProjectName_SampleID_R1.fastq.gz
ProjectName_SampleID_R2.fastq.gz
The filenames from Salus platform are handled as below:
<project_name>_<sampleID>_<read_#>.fastq.gz
Project name, sampleID and read # are used to match the files correctly.
You can alter the project name and sampleID fields without using space, punctuation and underscore (_) characters.
Following this naming convention, you can upload multiple samples (each with multiple fastq.gz files).
Please check the number of samples and matched files on the confirmation screen.
# Onso (Pacific Biosciences)
You can upload multiple samples with two or more files. For batch uploads, we only support filenames following the “Obc2fastq v6.0 naming convention” e.g.
ABCD123_L01_R1_Sample_Information.fastq.gz
ABCD123_L01_R2_Sample_Information.fastq.gz
<Flowcell_id>_<LaneSpec>_<ReadSpec|IndexSpec>_<Sample_id>.fastq.gz
FlowcellID, SampleID and ReadSpec|IndexSpec are used to match the files correctly. You can alter the FlowcellID and sampleID fields without using space or punctuation. Following this naming convention, you can upload multiple samples (each with multiple fastq.gz files).
Please check the number of samples and matched files on the confirmation screen.
# Advanced options
# Variant Calling parameters
A set of parameters is used to assess the quality of every variant called in a sample. Two parameters, the primary coverage threshold and the minimum alternative fraction threshold, can cause the classification of the variant as “FAILED”. The “FAILED” variant calls will not be displayed.
The variant calls with an alternative allele count less than the primary coverage threshold will be classified as “FAILED” and not be displayed.
The variant calls with alternative allele frequency less than the allele fraction threshold will be classified as “FAILED” and not be displayed.
# Other parameters
When calculating coverage metrics for the gene coverage and the kit’s on-target coverage percentages, SEQ uses four different thresholds. 1X and 5X are the preset values. The other two values may be customized by the user per upload.
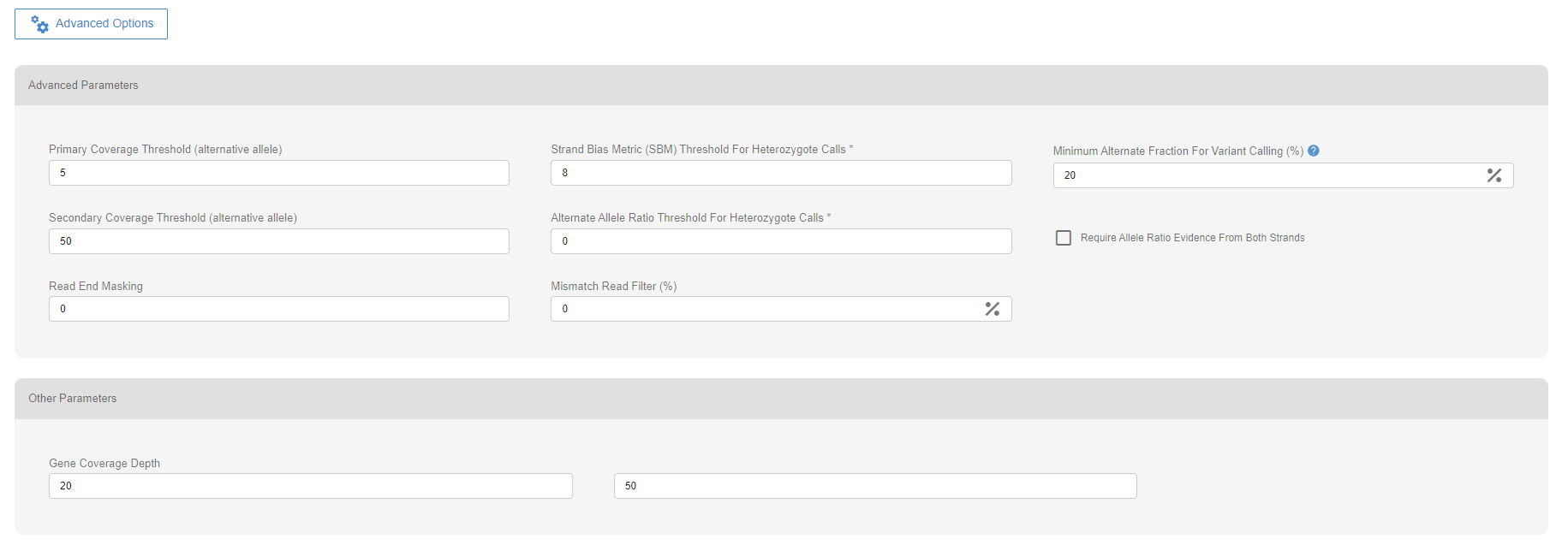
The default values of the advanced options are set under “Site Settings” in the Settings menu.
# Choose Analysis Version
SEQ has standard analysis versions pre-setup for every kit defined in the system. Data processing and variant calling are handled differently based on the sample type, sequencing platform, and selected analysis pipeline.
The SEQ platform has standard analysis versions pre-setup for every kit defined in the system. Calling variants are performed differently in different analysis versions.
# Analysis versions for Tumor/Normal Matched Samples (Capture based targeted panels, including WES)
| Name | Explanation | Alignment/ variant calling | BAM processing | MSI Detection | Available Genome Versions | Available Platforms |
|---|---|---|---|---|---|---|
| Sentieon BWA-TNhaplotyper2- somatic | Optimized for Exome Samples. | Sentieon BWA / TNhaplotyper2 | MarkDuplicate | MSIsensor (opens new window) | hg38 | Illumina MGI Element GeneMind Onso Salus |
| Sentieon BWA-TNScope- somatic | Optimized for capture-based somatic kits. | Sentieon BWA / TNScope | MarkDuplicate | MSIsensor (opens new window) | hg38 | Illumina MGI Element GeneMind Onso Salus |
# Analysis versions for Tumor Only Samples (Capture based targeted panels, including WES)
| Name | Explanation | Alignment/ variant calling | BAM processing | Fusion Calling | MSI Detection | Primer Trimming | Available Genome Versions | Available Platforms |
|---|---|---|---|---|---|---|---|---|
| BWA-Freebayes-PCR Dedup-Indel Realignment- somatic | Optimized for capture-based somatic kits. | BWA / Freebayes | Indel Realignment | RNA (STAR-Fusion) | msisensor2 (opens new window) | N/A | hg38 | Illumina MGI Element GeneMind Onso Salus |
| Sentieon (Solid, UMI) BWA-TNscope- somatic | Optimized for somatic UMI kits with with solid samples. | Sentieon BWA / TNScope | MarkDuplicate UMI Processing* | RNA (STAR-Fusion) | msisensor2 (opens new window) | N/A | hg38 | Illumina MGI Element GeneMind Onso Salus |
| Sentieon (ctDNA, UMI) BWA-TNscope- somatic | Optimized for somatic UMI kits with with ctDNA samples. | Sentieon BWA / TNScope | MarkDuplicate UMI Processing* | RNA (STAR-Fusion) | msisensor2 (opens new window) | N/A | hg38 | Illumina MGI Element GeneMind Onso Salus |
| Roche (Solid, UMI)-BWA-VarDict-RNA Fusion Calling- somatic | Optimized for Roche somatic UMI kits with solid samples. | BWA / VarDict | UMI Processing* | RNA (STAR-Fusion) | msisensor2 (opens new window) | N/A | hg38 | Illumina MGI Element GeneMind Onso Salus |
| Roche (cfDNA, UMI)-BWA-VarDict-RNA Fusion Calling- somatic | Optimized for Roche somatic UMI kits with cfDNA samples. | BWA / VarDict | UMI Processing* | RNA (STAR-Fusion) | msisensor2 (opens new window) | N/A | hg38 | Illumina MGI Element GeneMind Onso Salus |
| Nanodigmbio (Solid, UMI)-BWA-VarDict-RNA Fusion Calling- somatic | Optimized for Nanodigmbio somatic UMI kits with solid samples. | BWA / VarDict | UMI Processing* | RNA (STAR-Fusion) | msisensor2 (opens new window) | N/A | hg38 | Illumina MGI Element GeneMind Onso Salus |
| Nanodigmbio (cfDNA, UMI)-BWA-VarDict-RNA Fusion Calling- somatic | Optimized for Nanodigmbio somatic UMI kits with cfDNA samples. | BWA / VarDict | UMI Processing* | RNA (STAR-Fusion) | msisensor2 (opens new window) | N/A | hg38 | Illumina MGI Element GeneMind Onso Salus |
*the UMI barcode sequence is also required. For assistance, please contact support.
# Analysis versions for Tumor Only Samples (Amplicon based panels)
| Name | Explanation | Alignment/ variant calling | BAM processing | Fusion Calling | MSI Detection | Primer Trimming | Available Genome Versions | Available Platforms |
|---|---|---|---|---|---|---|---|---|
| BWA-Freebayes-BamKeser-Indel Realignment- somatic | Optimized for amplicon based somatic kits. | BWA / Freebayes | Indel Realignment | RNA (STAR-Fusion) | - | BamKeser* | hg38 | Illumina MGI Element GeneMind Onso Salus |
| BWA-Freebayes-BamKeser-Indel Realignment-Long Indel Finder- somatic | Optimized for amplicon-based somatic kits. Performs an additional step for long indel alterations. | BWA / Freebayes | Indel Realignment | RNA (STAR-Fusion) | - | BamKeser* | hg38 | Illumina MGI Element GeneMind Onso Salus |
*BamKeser is our in-house designed and precisely working primer trimming tool.
# Submit your data
As the last step, you can then click the "Upload" button to see the number of analyses and the list of files matched for each analysis. Please be sure that both of these pieces of information are correct and hit "Approve" to start the upload process or "Cancel" to make changes.
When you start the upload, you will see the progress for each file. Transferred samples will immediately begin processing without waiting for the entire batch to finish uploading.
SEQ Platform's upload process is secure and performs a checksum to ensure the files are transferred correctly. Please do not close the browser tab or shut down your computer. Also, please ensure that your computer will not go into sleep/hibernation mode during the upload. Otherwise, the upload process will be aborted. Our upload process is resistant to intermittent loss of internet connection.
When the upload process is completed, you will be redirected to the corresponding Run's page, and your samples will be queued for analysis. Refresh the corresponding Run page to see the last status of the analysis.
# Small Variant Detection
The variant caller used depends on the analysis version selected during upload — see Choose Analysis Version for the full list of available pipelines per platform and kit type.
| Sample Type | Variant Caller |
|---|---|
| Tumor/Normal matched (capture-based, incl. WES) | TNhaplotyper2, TNScope |
| Tumor-only (capture-based, incl. WES) | Freebayes, TNScope, VarDict (UMI-based kits) |
| Tumor-only (amplicon-based) | Freebayes |
# IMPORTANT NOTE: POST-PROCESSING
After variant calling, the Genomize SEQ platform processes the resulting VCF file to form a Genomize standard VCF file, which can be downloaded through the platform. The Genomize standard VCF line will have GSTD=1 in the info field. Standardization of the VCF file includes the following important steps:
- Minimal variant representation: Some callers produce redundant bases at the left-hand or right-hand side of either alternative or reference allele. This redundancy has to be removed to obtain the correct annotation of variants in the subsequent steps.
# RNA Fusion Detection
The RNA Fusion Detection pipeline identifies candidate gene fusion events from RNA sequencing (RNA-Seq) data through sequential preprocessing, alignment, and fusion discovery steps summarized in the table below.
| Step | Explanation | Tools |
|---|---|---|
| Adapter/Quality Trimming | Removes sequencing adapters, low-quality bases, and short reads to produce high-quality trimmed FASTQ files. | fastp (opens new window) |
| Alignment and Chimeric Read Detection | Aligns non-rRNA reads to the human genome and identifies chimeric junctions suggestive of fusion transcripts. | STAR (opens new window) |
| Fusion Transcript Detection | Analyzes chimeric junction data against the CTAT genome library* to detect and report candidate gene fusion events. | STAR-Fusion (opens new window) |
*The CTAT genome library is a publicly available reference genome library for RNA-Seq analysis (for the details, please refer to CTAT (opens new window)).
# TMB (Tumor Mutation Burden) Calculation
Tumor Mutational Burden is defined as non-synonymous SNP mutations per megabase.
If the covered region length is less than 1.1 megabases, TMB cannot be calculated and will be seen as 'N/A'.
For tumor only and ctDNA samples, TMB is calculated by the total number of non-synonymous and unique/novel SNP variants found in the sample, divided by the covered region on CDS. For tumor/matched-normalsamples, the variants detected in the normal sample are used to eliminate germline mutations.
Please see the table below for detailed parameters used for TMB calculation.
| Parameters | Tumor Only | T/N match | ctDNA |
|---|---|---|---|
| Minimum Allowed Covered Length | 1.1mb | 1.1mb | 1.1mb |
| Minimum Depth | 50 | 50 | 1000 |
| Minimum Allele Fraction | 0.05 | 0.05 | 0.002 |
| Maximum Allele Fraction | 0.90 | - | 0.90 |
| Population Database Allele Count | 50 | - | 50 |
| dbSNP filter | Yes | - | Yes |
| Chromosome Filter | MT | MT | MT |
| Filter Non-coding Regions | Yes | Yes | Yes |
| Filter MNVs | Yes | Yes | Yes |
| Filter No PASS Variants | Yes | Yes | Yes |
| Use SNV and Indels | Yes | Yes | Yes |
# MSI (Microsatellite Instability) Calculation
For MSI detection, MSIsensor (Niu et al., 2014 (opens new window)) is used on tumor–normal matched samples, while msisensor2 (opens new window) is applied to tumor-only samples.
# VCF Upload
# Run Selection
You can upload your samples to a new run by selecting “Create New Run” under “Run Name” and giving it a name on the “Name For New Run” field. You can also upload your samples to an existing run by selecting the previous run from the dropdown menu.
# Diagnostics
Next, you can add the diagnostic information including cancer type and the biomarker status.
# Cancer Type
- (Mandatory) Cancer Type (e.g. Breast Cancer)
- (Optional) Advanced Solid Tumor: Select this option if the sample or case involves a late-stage or metastatic solid tumor, regardless of the specific cancer type.
# Biomarkers
- (Optional) TMB status: tumor mutation burden status (high | intermediate | low).The specified TMB status will be used in associating relevant clinical trials and drugs.
- (Optional) MSI status: Microsatellite instability status ( high | low | stable ). The specified MSI status is displayed in the report and used in associating relevant clinical trials and drugs.
- (Optional) HRD status: Homologous Recombination Deficiency status (high | low). The HRD status will be used in associating relevant clinical trials and drugs.
- (Optional) TILs status: Tumor Infiltrating Lymphocytes status (high | low). The TILs status will be used in associating relevant clinical trials and drugs.
- (Optional) PD-L1 status: PD-L1 (CD274) expression status (positive | negative). The PD-L1 (CD274) status will be used in associating relevant clinical trials and drugs.
- (Optional) HER2 status: HER2 (ERBB2) expression status (positive | negative). HER2 (ERBB2) status will be used in associating relevant clinical trials and drugs.
# Select the VCF Files to be Uploaded
You can upload VCF files for SNVs from DRAGEN or other similar tools (See Supported Variant Callers) for the same sample in any combination you choose. VCF files should have vcf.gzextension. Files are matched using the sample info field, not the file names. Multisample VCFs are not supported.
# Submit your data
As the last step, you can then click the "Upload" button to see the number of analyses and the list of files matched for each analysis. Please be sure that both of these pieces of information are correct and hit "Approve" to start the upload process or "Cancel" to make changes.
When you start the upload, you will see the progress for each file. Transferred samples will immediately begin processing without waiting for the entire batch to finish uploading.
SEQ Platform's upload process is secure and performs a checksum to ensure the files are transferred correctly. Please do not close the browser tab or shut down your computer. Also, please ensure that your computer will not go into sleep/hibernation mode during the upload. Otherwise, the upload process will be aborted. Our upload process is resistant to intermittent loss of internet connection.
When the upload process is completed, you will be redirected to the corresponding Run's page, and your samples will be queued for analysis. Refresh the corresponding Run page to see the last status of the analysis.
# Supported Variant Callers
| Upload Type | Variant Types | Supported Callers | File Format (Extension) | Multisample Support |
|---|---|---|---|---|
| Small Variant | SNV INDEL | DeepVariant Dragen Freebayes GATK - Haplotype Caller Ion Torrent Variant Caller Isaac Variant Caller Mutect2 Pivat Sentieon DNAscope, TNScope VarDict Clair3 Qiagen CLC | VCF (.vcf.gz) | No |
# Unsupported Variant Callers and VCF Version Compatibility
Please note that using unlisted or unsupported variant callers may result in inaccurate VCF metrics. Unpredicted callers are categorized as "other," which may limit the capture of certain metrics. In some cases, custom integration may be required to support these callers fully. Only VCF version 4.1 or newer is supported for Copy Number, Structural and Short Tandem VCF files. If issues persist or critical data appears missing, please contact support for assistance.
# Mitochondrial Variants
The Revised Cambridge Reference Sequence (rCRS, NC_012920.1) is used as the reference for the mitochondrial genome regardless of the genome version used in VCF generation, which is the recommended sequence for clinical use (McCormick et al., 2020 (opens new window)). If the VCF file contains variants called using the older Yoruban (YRI) mitochondrial reference genome, errors may result due to incompatibility with our annotation sources. Unsupported chrM variants should also be removed before upload to prevent genome compatibility issues. For assistance, or if issues arise, please contact support.
# Filtering Parameters Applied to Small Variants
No Call Filter: Small variants with a 'no call' status are excluded, ensuring that only fully determined genotypes are included in the analysis.
Chromosome Filter: Small variants that are not on chromosomes 1-22, X,M are filtered out.
# Cloud Browser
To use the cloud browser, select or create your run, select the sequencing platform and the kit by following the directions above. After the kit selection, you will see the option to select either your “COMPUTER” or the “CLOUD BROWSER” as the data source.
When you select the "CLOUD BROWSER" option, click the PLUS (➕) button to open the cloud browser interface. Using the cloud browser, you can choose the files with which you want to start the analysis and click “DONE”. The rest of the process is the same as described above. Please note that there is a 3-minute duration between each cloud upload process, and files will be removed from your cloud account upon starting the analysis.